Accelerate Biological Research
info@kendallscientific.com
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-617.299.7367 (Int’l)
+1-888.733.6849
+1-617.299.7367 (Int’l)
+1-617.299.7367 (Int’l)
For quotations, please use our online quotation form, and you may also contact us by
service@kendallscientific.com
+1-888.733.6849 (Toll-free)
+1-617.299.7367 (Int’l))
+1-888.733.6849
Our customer service representatives are available 24 hours, Monday through Friday to assist you.| Reactivity | Human Mouse |
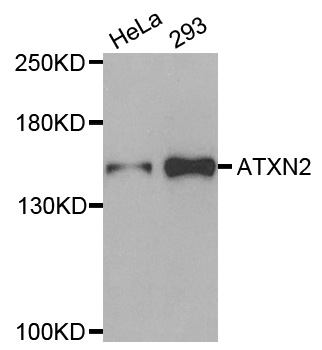
| Tested applications | WB |
| Recommended Dilution | WB 1:500 - 1:2000 |
| Calculated MW | 140kDa |
| Observed MW | Refer to Figures |
| Immunogen | A synthetic peptide of human ATXN2 |
| Storage Buffer | Store at 4℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Synonym | ATX2; SCA2; ASL13; TNRC13; |
The autosomal dominant cerebellar ataxias (ADCA) are a heterogeneous group of neurodegenerative disorders characterized by progressive degeneration of the cerebellum, brain stem and spinal cord. Clinically, ADCA has been divided into three groups: ADCA types I-III. Defects in this gene are the cause of spinocerebellar ataxia type 2 (SCA2). SCA2 belongs to the autosomal dominant cerebellar ataxias type I (ADCA I) which are characterized by cerebellar ataxia in combination with additional clinical features like optic atrophy, ophthalmoplegia, bulbar and extrapyramidal signs, peripheral neuropathy and dementia. SCA2 is caused by expansion of a CAG repeat in the coding region of this gene. This locus has been mapped to chromosome 12, and it has been determined that the diseased allele contains 37-50 CAG repeats, compared to 17-29 in the normal allele. Longer expansions result in earlier onset of the disease. Alternatively spliced transcript variants encoding different isoforms have been identified but their full length sequence has not been determined.
N/A



Copyrights © 2014 Kendall Scientific, 250 Parkway Drive, Suite 150 Lincolnshire, IL 60069